Control statements¶
So far we have discussed:
- Expressions, including function calls
- Assignments
- Augmented assignments
- Various forms of
import - Assertions
returnyield(to implement generators)pass- statements:
def- for defining functionswith- to use with files
We will discuss about compound statements:
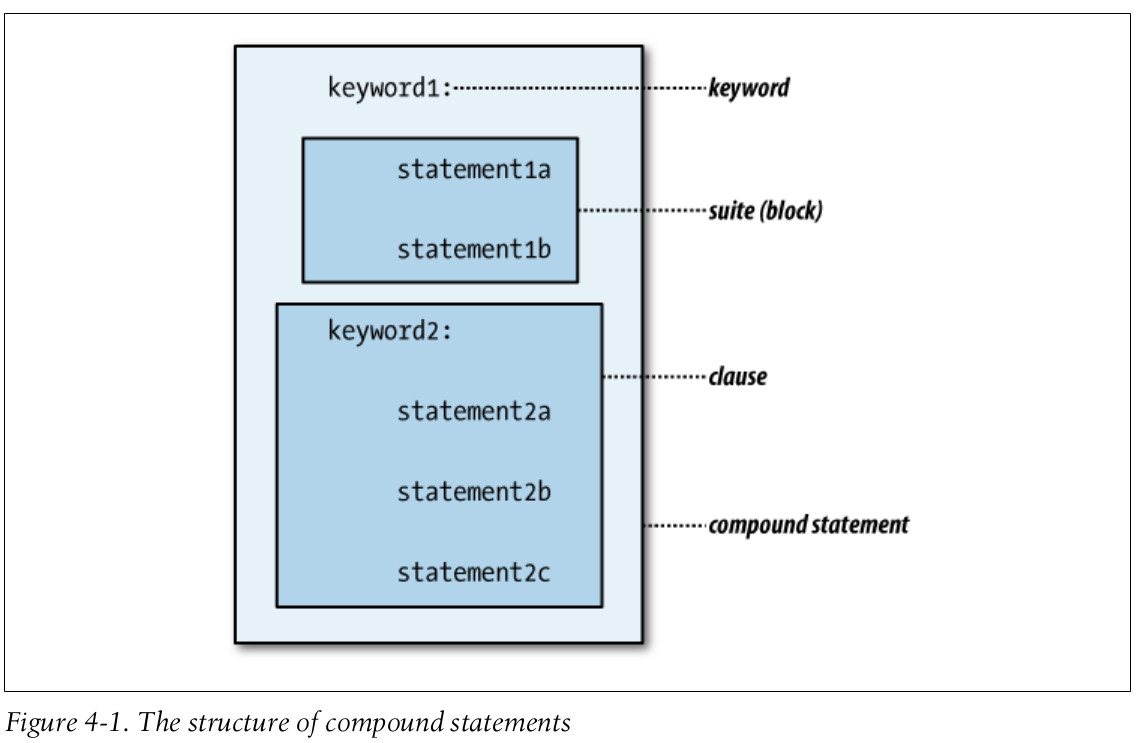
The four kinds of compound statements introduced here are:
- Conditionals
- Loops
- Iterations
- Exception handlers
Conditionals¶
Conditionals in Python are compound statements beginning with if.
if expression: statements
- During the import of a module
__name__is bound to the name of the module, but while the file is being executed__name__is bound to '__main__' . - This gives you a way to include statements in your Python files that are executed only when the module is run or, conversely, only when it is imported.
def do_tests():
pass
if __name__ == '__main__':
do_tests()
One-Alternative Conditionals¶
if expression:
statements1
else:
statements2
if __name__ == '__main__':
do_tests()
else:
print(__name__,'has been imported')
Multi-Test Conditionals¶
if expression1:
statements1
elif expression2:
statements2
...any number of additional elif clauses
else:
statements
Loops¶
A loop is a block of statements that gets executed as long as some condition is true. Statements get executed if the test expresssion is satisfied.
while expression:
statements- If code is to be execute once the test expression is false
while expression: statements1 else: statments2
- Two simple statements associated eith loops and iterations:
continueandbreak - The
continuestatement causes execution of the loop to return to the test. - The
breakstatement interrupts the entire loop’s execution, causing the program to continue with the statement following the while .
def echo():
"""Echo the user's input until an empty line is entered"""
while echo1():
pass
def echo1():
"""Prompt the user for a string, "echo" it, and return it"""
line = input('Say something: ')
print('You said "', line, '"', sep='')
return line
echo()
def polite_echo():
"""Echo the user's input until it equals 'bye'"""
while echo1() != 'bye':
pass
polite_echo()
def recording_echo():
"""Echo the user's input until it equals 'bye', then return a
list of all the inputs received"""
lst = [] # initialize entry and lst
entry = echo1() # get the first input
while entry != 'bye': # test entry
lst.append(entry) # use entry
entry = echo1() # change entry
# ... repeat
return lst # return result
recording_echo()
Looping forever¶
initialize values
while True:
# change values
if test values:
return
use values
# repeat
return resultdef recording_echo_with_conditional():
"""Echo the user's input until it equals 'bye', then return a
list of all the inputs received"""
seq = []
# no need to initialize a value to be tested since nothing is tested!
while True:
entry = echo1()
if entry == 'bye':
return seq
seq.append(entry)
recording_echo_with_conditional()
RNA_codon_table = {
# Second Base
# U C A G
# U
'UUU': 'Phe', 'UCU': 'Ser', 'UAU': 'Tyr', 'UGU': 'Cys', # UxU
'UUC': 'Phe', 'UCC': 'Ser', 'UAC': 'Tyr', 'UGC': 'Cys', # UxC
'UUA': 'Leu', 'UCA': 'Ser', 'UAA': '---', 'UGA': '---', # UxA
'UUG': 'Leu', 'UCG': 'Ser', 'UAG': '---', 'UGG': 'Urp', # UxG
# C
'CUU': 'Leu', 'CCU': 'Pro', 'CAU': 'His', 'CGU': 'Arg', # CxU
'CUC': 'Leu', 'CCC': 'Pro', 'CAC': 'His', 'CGC': 'Arg', # CxC
'CUA': 'Leu', 'CCA': 'Pro', 'CAA': 'Gln', 'CGA': 'Arg', # CxA
'CUG': 'Leu', 'CCG': 'Pro', 'CAG': 'Gln', 'CGG': 'Arg', # CxG
# A
'AUU': 'Ile', 'ACU': 'Thr', 'AAU': 'Asn', 'AGU': 'Ser', # AxU
'AUC': 'Ile', 'ACC': 'Thr', 'AAC': 'Asn', 'AGC': 'Ser', # AxC
'AUA': 'Ile', 'ACA': 'Thr', 'AAA': 'Lys', 'AGA': 'Arg', # AxA
'AUG': 'Met', 'ACG': 'Thr', 'AAG': 'Lys', 'AGG': 'Arg', # AxG
# G
'GUU': 'Val', 'GCU': 'Ala', 'GAU': 'Asp', 'GGU': 'Gly', # GxU
'GUC': 'Val', 'GCC': 'Ala', 'GAC': 'Asp', 'GGC': 'Gly', # GxC
'GUA': 'Val', 'GCA': 'Ala', 'GAA': 'Glu', 'GGA': 'Gly', # GxA
'GUG': 'Val', 'GCG': 'Ala', 'GAG': 'Glu', 'GGG': 'Gly' # GxG
}
def translate_RNA_codon(codon):
"""RNA codon lookup from a dictionary"""
return RNA_codon_table[codon]
def aa_generator(rnaseq):
"""Return a generator object that produces an amino acid by
translating the next three characters of rnaseq each time next is
called on it"""
return (translate_RNA_codon(rnaseq[n:n+3])
for n in range(0, len(rnaseq), 3))
def translate(rnaseq):
"""Translate rnaseq into amino acid symbols"""
gen = aa_generator(rnaseq)
seq = ''
aa = next(gen, None)
while aa:
seq += aa
aa = next(gen, None)
return seq
translate('AAUAAGCGCGGGUAUGAUGCGGUCUUG')
def read_sequence(filename):
"""Given the name of a FASTA file named filename, read and return
its first sequence, ignoring the sequence's description"""
seq = ''
with open(filename) as file:
line = file.readline()
while line and line[0] == '>':
line = file.readline()
while line and line[0] != '>': # must check for end of file
seq += line
line = file.readline()
return seq
read_sequence('data/aa003.fasta')
Iterations¶
- Doing something to each element of a collection is called iteration.
- Iteration statements all begin with the keyword
for.for item in collection: do something with item
File Iteration¶
with open(filename) as file:
for line in file:
do something with lineDictionary Iteration¶
for key in dictionary.keys():
do something with keyfor key in dictionary.values():
do something with valuefor key, value in dictionary.items():
do something with key and valueNumbering Iterations¶
Use the function enumerate and tuple unpacking to generate numerical keys in parallel with the values in an iterable.
for n, value in enumerate(iterable):
do something with n and valuea = list(range(10,20))
for n, value in enumerate(a):
print(n, value, sep='\t')
def print_collection(collection):
for item in collection:
print(item)
print()
collection = [2, 3, 4, 5]
[print(item) for item in collection]
Repeat - Template¶
To repeat a block of statements n times, iterate over range(n).
for count in range(n):
statementsFrequently, count would not even be used in the body of the iteration.
Collect - Template¶
- A Collect iteration starts with an empty collection and uses a method or operator appropriate to its type to add something to it for each iteration.
result = [] for item in collection: statements using item result.append(expression based on the statements) return result
#Reading FASTA entries with a Collect iteration
def read_FASTA_iteration(filename):
sequences = []
descr = None
with open(filename) as file:
for line in file:
if line[0] == '>':
if descr: # have we found one yet?
sequences.append((descr, seq))
descr = line[1:-1].split('|')
seq = '' # start a new sequence
else:
seq += line[:-1]
sequences.append((descr, seq)) # add the last one found
return sequences
def read_FASTA(filename):
with open(filename) as file:
return [(part[0].split('|'),
part[2].replace('\n', ''))
for part in
[entry.partition('\n')
for entry in file.read().split('>')[1:]]]
def read_FASTA_loop(filename):
sequences = []
descr = None
with open(filename) as file:
line = file.readline()[:-1] # always trim newline
while line:
if line[0] == '>':
if descr: # any sequence found yet?
sequences.append((descr, seq))
descr = line[1:].split('|')
seq = '' # start a new sequence
else:
seq += line
line = file.readline()[:-1]
sequences.append((descr, seq)) # easy to forget!
return sequences
def test():
filename = 'data/aa003.fasta'
result1 = read_FASTA_iteration(filename)
result2 = read_FASTA(filename)
result3 = read_FASTA_loop(filename)
assert result1 == result2 == result3
print('All tests passed')
test()
Combine - Template¶
- Sometimes we want to perform an operation on all of the elements of a collection to yield a single value.
def product(coll):
"""Return the product of the elements of coll converted to floats, including elements that
are string representations of numbers; if coll has an element that is a string but doesn't
represent a number, an error will occur"""
result = 1.0 # initialize
for elt in coll:
result *= float(elt) # combine element with
return result # accumulated result
product(range(1, 6))
### Combine: identifying the longest FASTA sequence
def longest_sequence(filename):
longest_seq = ''
for info, seq in read_FASTA(filename):
longest_seq = max(longest_seq, seq, key=len)
return longest_seq
longest_sequence('data/aa003.fasta')
Count¶
- A Count iteration “combines” the value 1 for each element of the iteration.
count = 0 for item in iterable: count += 1 return count
Collection Combine¶
- In this variation on Combine, an action is performed on each element of a collection that produces a collection as a result, but instead of returning a collection of the results, the iteration combines the results into (reduces the results to) a single collection.
result = [] for item in collection: result += fn(item) # merge result with previous results return result
### Collection Combine: sequence IDs from multiple files
def extract_gi_id(description):
"""Given a FASTA file description line, return its GenInfo ID if it has one"""
if description[0] != '>':
return None
fields = description[1:].split('|')
if 'gi' not in fields:
return None
return fields[1 + fields.index('gi')]
def get_gi_ids(filename):
"""Return a list of the GenInfo IDs of all sequences found in the file named filename"""
with open(filename) as file:
return [extract_gi_id(line) for line in file if line[0] == '>']
def get_gi_ids_from_files(filenames):
"""Return a list of the GenInfo IDs of all sequences found in the files whose names are
contained in the collection filenames"""
idlst = []
for filename in filenames:
idlst += get_gi_ids(filename)
return idlst
def test():
filenames = ('data/nadh.fasta',
'data/aa003.fasta',
'data/BacillusSubtilisPlastmidP1414.fasta'
)
print(get_gi_ids_from_files(filenames))
test()
Search¶
- Search is a simple variation on Do:
for item in collection: if test item: return item
### Extracting a sequence by ID from a large FASTA file
def FASTA_search_by_gi_id(id, file):
for line in file:
if (line[0] == '>' and str(id) == get_gi_id(line)):
return read_FASTA_sequence(file)
def search_FASTA_file_by_gi_id(id, filename):
"""Return the sequence with the GenInfo ID ID from the FASTA file
named filename, reading one entry at a time until it is found"""
id = str(id) # user might call with a number
with open(filename) as file:
return FASTA_search_by_gi_id(id, file)
def read_FASTA_sequence(file):
seq = ''
for line in file:
if not line or line[0] == '>':
return seq
seq += line[:-1]
def get_gi_id(description):
fields = description[1:].split('|')
if fields and 'gi' in fields:
return fields[(1 + fields.index('gi'))]
filename = 'data/Acidobacterium-capsulatum-coding-regions.fasta'
search_FASTA_file_by_gi_id('225793573', filename)
def rna_sequence_is_valid(seq):
for base in seq:
if base not in 'UCAGucag':
return False
return True
def dna_sequence_contains_N(seq):
for base in seq:
if base == 'N':
return True
Filter¶
Filtering is similar to searching, but instead of returning a result the first time a match is found, it does something with each element for which the match was successful.
Filtered Do¶
- A Filtered Do performs an action for each item that meets a specified condition.
for item in collection: if test item: statements using item
### Printing the header lines from a FASTA file
def print_FASTA_headers(filename):
with open(filename) as file:
for line in file:
if line[0] == '>':
print(line[1:-1])
print_FASTA_headers('data/aa010.fasta')
Filtered Collect¶
- Here, the values for which test is true are collected one at a time.
result = [] for item in collection: if test item: statements using item result.append(expression based on the statements) return result
### Extracting sequences with matching descriptions
def extract_matching_sequences(filename, string):
"""From a FASTA file named filename, extract all sequences whose descriptions contain string"""
sequences = []
seq = ''
with open(filename) as file:
for line in file:
if line[0] == '>':
if seq: # not first time through
sequences.append(seq)
seq = '' # next sequence detected
includeflag = string in line # flag for later iterations
else:
if includeflag:
seq += line[:-1]
if seq: # last sequence in file is included
sequences.append(seq)
return sequences
for seq in extract_matching_sequences('data/aa010.fasta', 'factor'):
print(seq)
Filtered Combine¶
### Filtered Combine: another definition of product
def is_number(value):
"""Return True if value is an int or a float"""
return isinstance(value, int) or isinstance(value, float)
def product(coll):
"""Return the product of the numeric elements of coll"""
result = 1.0 # initialize
for elt in coll:
if is_number(elt):
result = result * float(elt) # combine element with accumulated result
return result
product((2, None, 3.5, 'four'))
Filtered Count¶
count = 0
for item in iterable:
if test item:
count += 1
return countNested Iterations¶
for outer in outer_collection:
for inner in inner_collection:
do something with inner and outer### A nested iteration
def list_sequences_in_files(filelist):
"""For each file whose name is contained in filelist, list the
description of each sequence it contains"""
for filename in filelist:
print(filename)
with open(filename) as file:
for line in file:
if line[0] == '>':
print('\t', line[1:-1])
filenames = ('data/nadh.fasta',
'data/aa003.fasta',
'data/BacillusSubtilisPlastmidP1414.fasta'
)
list_sequences_in_files(filenames)
### A two-function nested iteration
def list_sequences_in_files(filelist):
"""For each file whose name is contained in filelist, list the
description of each sequence it contains"""
for filename in filelist:
print(filename)
with open(filename) as file:
list_sequences_in_file(file)
def list_sequences_in_file(file):
for line in file:
if line[0] == '>':
print('\t', line[1:-1])
filenames = ('data/nadh.fasta',
'data/aa003.fasta',
'data/BacillusSubtilisPlastmidP1414.fasta'
)
list_sequences_in_filesb(filenames)
### Printing the codon table
RNA_codon_table = {
# Second Base
# U C A G
# U
'UUU': 'Phe', 'UCU': 'Ser', 'UAU': 'Tyr', 'UGU': 'Cys', # UxU
'UUC': 'Phe', 'UCC': 'Ser', 'UAC': 'Tyr', 'UGC': 'Cys', # UxC
'UUA': 'Leu', 'UCA': 'Ser', 'UAA': '---', 'UGA': '---', # UxA
'UUG': 'Leu', 'UCG': 'Ser', 'UAG': '---', 'UGG': 'Urp', # UxG
# C
'CUU': 'Leu', 'CCU': 'Pro', 'CAU': 'His', 'CGU': 'Arg', # CxU
'CUC': 'Leu', 'CCC': 'Pro', 'CAC': 'His', 'CGC': 'Arg', # CxC
'CUA': 'Leu', 'CCA': 'Pro', 'CAA': 'Gln', 'CGA': 'Arg', # CxA
'CUG': 'Leu', 'CCG': 'Pro', 'CAG': 'Gln', 'CGG': 'Arg', # CxG
# A
'AUU': 'Ile', 'ACU': 'Thr', 'AAU': 'Asn', 'AGU': 'Ser', # AxU
'AUC': 'Ile', 'ACC': 'Thr', 'AAC': 'Asn', 'AGC': 'Ser', # AxC
'AUA': 'Ile', 'ACA': 'Thr', 'AAA': 'Lys', 'AGA': 'Arg', # AxA
'AUG': 'Met', 'ACG': 'Thr', 'AAG': 'Lys', 'AGG': 'Arg', # AxG
# G
'GUU': 'Val', 'GCU': 'Ala', 'GAU': 'Asp', 'GGU': 'Gly', # GxU
'GUC': 'Val', 'GCC': 'Ala', 'GAC': 'Asp', 'GGC': 'Gly', # GxC
'GUA': 'Val', 'GCA': 'Ala', 'GAA': 'Glu', 'GGA': 'Gly', # GxA
'GUG': 'Val', 'GCG': 'Ala', 'GAG': 'Glu', 'GGG': 'Gly' # GxG
}
### RNA codon lookup from a dictionary
def translate_RNA_codon(codon):
return RNA_codon_table[codon]
DNA_bases = ('T', 'C', 'A', 'G')
def translate_DNA_codon(codon):
return DNA_codon_table[codon]
def print_codon_table():
"""Print the DNA codon table in a nice, but simple,
arrangement"""
for base1 in DNA_bases: # horizontal section (or "group")
for base3 in DNA_bases: # line (or "row")
for base2 in DNA_bases: # vertical section (or "column")
# the base2 loop is inside the base3 loop!
print(base1+base2+base3,
translate_DNA_codon(base1+base2+base3),
end=' ')
print()
print()
print_codon_table()